Genetic Testing for Hereditary Thoracic Aortic Aneurysm Disease
Hereditary Thoracic Aortic Aneurysm Disease (HTAD) is a group of genetic conditions that involve abnormalities in the aorta, the major blood vessel in the chest (thoracic) that carries blood from the heart to the body (Figure 1). If you have HTAD, you are at increased risk to develop progressive enlargement of the aorta, called an aneurysm (Figure 2), as well as at risk for an aortic dissection (a tear in the aortic wall) (Figure 3), which can be fatal. HTAD often runs in families and affects both males and females. HTAD can occur in multiple generations in your family, as the genes associated with HTAD can be passed (inherited) from your parents or from you to your children (Figure 4).
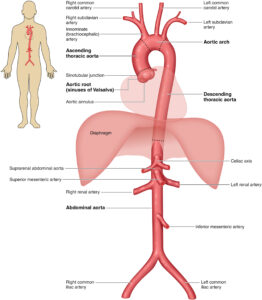
Figure 1 – The Anatomy of the Aorta and Its Main Branches (Source: 2022 ACC/AHA Guideline for the Diagnosis and Management of Aortic Disease).
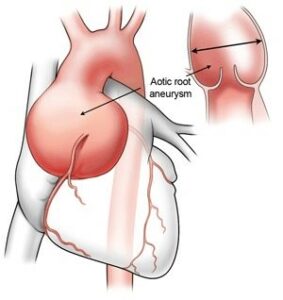
Figure 2 – Aortic root aneurysm (Source: https://www.heart-valve-surgery.com/heart-surgery-blog/2014/08/04/enlarged-aorta-normal-size-after-bicuspid-aortic-valve-replacement/)
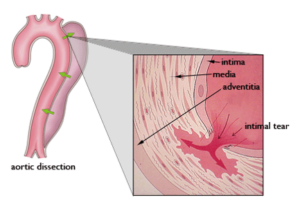
Figure 3 – Aortic Dissection
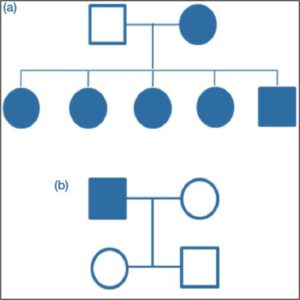
Figure 4 – Genetic Pedigree of a Family with Marfan Syndrome
HTAD may be syndromic or non-syndromic. A syndrome is a collection of abnormalities in the same person. Some of the syndromic HTAD are hereditary connective tissue disorders. Connective tissue supports skin, bones, blood vessels, and many other organs and tissues in the body.
People with a syndromic HTAD are at risk for aneurysms of the thoracic aorta (the part of the aorta near the heart), and can also have other medical complications, including abnormalities of other parts of the aorta (abdominal aorta and its branches) or other heart/blood vessel problems, as well as complications including vision, joint, bone, neurological and skin abnormalities. Examples of syndromic HTADs include Marfan syndrome, Loeys-Dietz syndrome, Vascular Ehlers-Danlos syndrome, and Shprintzen-Goldberg syndrome
Individuals with non-syndromic HTAD may not have risks for complications outside the aorta. In families it is often referred to as ‘Familial Thoracic Aortic Aneurysm (FTAA)’ or ‘Familial Thoracic Aortic Aneurysm Dissection (FTAAD)‘. Some FTAADs are also associated with aortic valve abnormalities (bicuspid aortic valve), congenital heart conditions (atrial septal defect, patent ductus arteriosus), cerebral (brain aneurysms) and/or changes in the skin where there is a lacy pattern of the blood vessels called livedo reticularis (as may be present in ACTA2 gene-related HTAD).
While the complications of HTAD can be life-threatening and scary, HTAD is treatable. Some of the most serious complications of HTAD may be lessened or prevented with regular medical care that includes heart and blood vessel imaging, timely aortic and blood vessel surgery, and other syndromic-HTAD care recommendations.
Regular aortic imaging (with an echocardiogram, MRA (magnetic resonance angiogram, using a magnetic field to generate images), or CTA (computerized tomography angiogram, using x-rays)) can track the size of the aorta over time. Aortic aneurysms do not often cause symptoms and so are not often diagnosed unless aortic imaging has been done. Aortic aneurysm often first displays symptoms after it has torn (dissected) and becomes an emergency situation of an aortic dissection. Treatment with certain medications may slow down aortic enlargement, and aortic replacement surgery can prevent a dissection by replacing the enlarged part of the aorta with a graft. The earlier an aneurysm or risk for aneurysm can be diagnosed, the better chance of avoiding the serious complications of HTAD.
A gene is a specific set of instructions present in the cells of the body. We have about 20,000 total genes, which provide all types of instructions for the proper development and function of your body organs and systems. Genes often work and interact with one another to keep our bodies running well.
Genes are made up of long stretches of molecules called DNA. DNA has its own special alphabet of letters (the DNA code), and each gene has a specific spelling for its instructions, often hundreds of thousands or even millions of letters long.
A difference in the typical spelling of a gene, even a change in one letter, can alter a gene’s instructions and lead to health problems. A difference in a gene that negatively affects that gene’s function is called a “(likely) pathogenic variant.”
There are many genes with instructions that are involved in the proper creation and functioning of connective tissue and/or blood vessels. These genes work together to create and regulate healthy, well-functioning tissues. A (likely) pathogenic variant in one gene disrupts not only that gene’s function, but often affects how other genes in the group function, leading to abnormal connective tissue and vessels, which lead to the features and medical issues in HTADs.
More than 20 genes have been identified in HTADs. Typically, only one of the HTAD genes contains a pathogenic variant in a specific person or family. The gene variant responsible for HTAD in a person/family is often unique in that family compared to other unrelated people with HTAD. In some cases, many families are found to have the same pathogenic variant in the gene causing the disease.
Certain genes are associated with specific syndromes (for example, variants in FBN1 gene cause Marfan syndrome; COL3A1 leads to Vascular EDS, variants in TGFBR1, TGFBR2, SMAD3, TGFB2, TGFB3, and SMAD2 lead to Loeys-Dietz syndrome), while other genes cause FTAAD. In some cases, however, the same gene can cause both a syndromic and a non-syndromic form of HTAD. For example, some people with a pathogenic variant in TGFBR1 have aneurysm disease and the physical features of Loeys-Dietz syndrome (LDS), while others with a different TGFBR1 pathogenic variant may have thoracic aortic aneurysm disease alone and no outward features of LDS.
Some of the most common HTAD and their associated conditions are listed in Table 1.
| Syndromic HTAD Genes | Condition | Clinical Features |
| FBN1 | Marfan syndrome | Aortic root aneurysm, aortic dissection, TAA, MVP, long bone overgrowth, arachnodactyly, dolichostenomelia, scoliosis, pectus deformities, ectopia lentis, myopia, tall stature, pneumothorax, dural ectasia |
| TGFBR1, TGFBR2, SMAD3, TGFB2, TGFB3 | Loeys-Dietz syndrome | TAA, branch vessel aneurysms, aortic dissection, arterial tortuosity, MVP, craniosynostosis, hypertelorism, bluish sclera, bifid/broad uvula, translucent skin, visible veins, club feet, dural ectasia, and premature osteoarthritis and peripheral neuropathy |
| COL3A1 | Vascular Ehlers-Danlos syndrome | AA, AAA, arterial rupture, aortic dissection, MVP, bowel and uterine rupture, pneumothorax, translucent skin, atrophic scars, small joint hypermobility, easy bruising, carotid-cavernous fistula |
| SLC2A10 | Arterial tortuosity syndrome | Tortuous large and medium sized arteries, aortic dilation, craniofacial, skin and skeletal features |
| SKI | Shprintzen-Goldberg syndrome | Craniosynostosis, skeletal features, aortic dilation |
| FLNA | Ehlers-Danlos syndrome with periventricular nodular heterotopia | X-linked, periventricular nodular heterotopia, TAA, BAV, MV disease, PDA, VSD, seizures, jointhypermobility |
| BGN | Meester-Loeys syndrome | X-linked, TAA, aortic dissection, MV disease |
| LOX | LOX-related TAA | TAA, BAV, aortic dissection, Marfanoid habitus in some |
| ACTA2 | Smooth muscle dysfunction syndrome | TAA, moyamoya-like cerebrovascular disease, pulmonary hypertension, pulmonary disease, hypoperistalsis, hypotonic bladder, congenital mydriasis |
| Nonsyndromic HTAD (Familial TAA) Genes | Condition | Clinical Features |
| ACTA2 | FTAA | TAA, aortic dissection, premature CAD and moyamoya-like cerebrovascular disease, livedo reticularis, iris flocculi |
| MYH11 | FTAA | TAA, aortic dissection, PDA |
| MYLK | FTAA | Aortic dissection at relatively small aortic size |
| PRKG1 | FTAA | Aortic dissection at young ages at small aortic sizes |
| MAT2A | FTAA | TAA, aortic dissection, BAV |
| MFAP5 | FTAA | TAA, aortic dissection, skeletal features may be present |
| FOXE3 | FTAA | TAA, aortic dissection |
| THSD4 | FTAA | TAA, aortic dissection |
| BAV-associated Ascending Aortic Aneurysm Genes | Condition | Clinical Features |
| NOTCH1 | Familial BAV/AS and TAA | Aortic valve stenosis, TAA |
| TGFBR2, MAT2A, GATA5, SMAD6, LOX, R0B04, TBX20 | BAV with TAA | Syndromic and nonsyndromic HTAD and FTAA with an increased frequency of BAV |
| X0, Xp | Turner syndrome | BAV, CoA, TAA, aortic dissection, short stature, lymphedema, webbed neck, premature ovarian failure |
Table 1 – Hereditable thoracic aortic diseases (HTAD) (Source: 2022 ACC/AHA Guideline for the Diagnosis and Management of Aortic Disease)
Most HTAD genes follow an inheritance pattern in a family called “autosomal dominant inheritance.” Everyone has two copies of each gene (one from mom, one from dad). Individuals who have a dominant variant in one gene, and a normal working copy of that gene on the other parent’s chromosome, will be affected by that condition despite the working copy. This means that each child of a person with HTAD has a 50% chance of inheriting the (likely) pathogenic variant putting them at risk to develop HTAD.
Therefore, if you have a (likely) pathogenic variant in an HTAD gene, it may be passed on to your children. Because HTAD conditions follow an autosomal dominant pattern of inheritance, there is a 50% chance of passing on the gene defect each time you have a child. This is because each time you have a child, there is a 50% chance the child will inherit the gene defect (and a 50% chance the child will inherit the normal gene). It is not possible to skip a generation.
Genetic testing is a medical test that can be performed by collecting cells from blood or saliva to look at DNA. Testing is done to see if there is a genetic change or variant in one of the HTAD genes that could increase the risk for development of an aortic aneurysm and dissection. If genetic testing is ordered by a medical provider, usually all of the HTAD genes are tested together on a panel. If the genetic diagnosis or variant has already been identified in a family member, your medical provider can order genetic testing for this specific genetic variant to see if other relatives are at risk.
It is not often possible to make the diagnosis of a specific HTAD by a physical examination alone or to rule out the condition by a physical examination alone. Therefore, genetic testing is very important for determining which gene defect is present and the best treatment.
There can be a lot of overlap in the outward features of these conditions. Genetic testing can identify the specific gene responsible for the condition. Identifying the specific gene responsible for the HTAD may help your doctors decide about your treatment, including when aortic surgery is recommended. Also, once a (likely) pathogenic variant in a gene has been identified as the cause in a family, all other family members can be tested to see if they have the same variant and are at risk for thoracic aortic aneurysm and aortic dissection.
Genetic test results may guide your treatment
The results of the testing can often help your doctor decide on the treatment and management for your HTAD. Examples include determining how often the aorta and its branches should be imaged, what type of imaging is used (echo, MRA, CTA), what type of medication and when to start it, and when to recommend aortic surgery to replace or repair an aneurysm to prevent a dissection. For those with syndromic HTADs, finding out about a variant provides important information about additional issues seen in the syndrome for which that a person needs to be monitored and treated.
Genetic test results can impact recommendations and care for your relatives
If a (likely) pathogenic variant has been identified, your family members can undergo genetic testing to see if they also have this variant. If any relatives test positive, they are recommended to begin screening for aneurysms and other abnormal blood vessels (as well as other features of the condition) with testing such as an echocardiogram, MRA, and CTA. Relatives who test negative are not at risk for HTAD and cannot pass on the risk to their children. Genetic testing can help diagnose HTAD in yourself and your family members and often allows for earlier identification of at-risk family members who could benefit from screening and treatment.
It is important to understand that genetic testing will not always identify a specific variant to explain the cause of the aneurysm disease in many families, even when there is a clear pattern of aortic aneurysm disease running in a family. Researchers are continuously finding new genetic variants to explain aneurysm disease. When genetic testing does not find a variant in a gene to explain the aneurysm condition, it is still important to screen first degree relatives (parents, siblings and children) with aortic imaging (echocardiogram, MRA or CTA) to see if anyone else in the family is has aortic enlargement.
It is generally recommended that the first person in a family to undergo HTAD genetic testing be someone who has the most features raising concern for an HTAD diagnosis, such as young individuals with aortic aneurysm/dissection and/or other strongly syndromic HTAD associated features (for example, an individual with dislocated eye lenses in Marfan syndrome).
There are generally three types of results from HTAD testing:
Positive – when a (likely) pathogenic variant is identified, providing or confirming a diagnosis of a specific HTAD. In HTAD, if the variant is identified in an affected family member, other family members can then be tested to see if they have this specific variant. Anyone who tests positive, regardless of symptoms, should follow the screening and management guidelines as recommended for their specific type of HTAD.
Negative – no (likely) pathogenic variant(s) in any of the genes tested are reported. It is important to understand that a negative genetic test does NOT rule out a diagnosis of HTAD, especially for the non-syndromic HTADs. Even in a family in which one recognizes that there are multiple family members with aneurysm disease, the chance of identifying the specific variant that causes a non-syndromic HTAD is only approximately 25%. Importantly, in a family with thoracic aortic aneurysm disease, even if genetic test results are negative, first-degree relatives (parents, siblings, and children) of a person with a suspected HTAD are recommended to have an echocardiogram, MRA, or CTA performed to check the aorta.
It is also important to check in with a geneticist, genetic counselor, or other knowledgeable physician if you had negative HTAD genetic testing in the past, as new genes are continuing to be identified that you may have not been tested for previously.
Variant of uncertain significance (VUS) – Spelling differences in genes are common, contributing to varying traits seen in people, but are not always harmful and they do not always cause syndromes or other health problems. Spelling changes in a gene with effects that we do not currently understand are called variants of unknown significance. VUS are often identified in genetic testing. Generally, it is not recommended to test unaffected family members (relatives without any physical signs of HTAD/normal aorta) for VUS. This is because results from VUS testing in an unaffected relative does not determine whether or not that relative is at risk to develop HTAD, nor if that relative should have echocardiogram or not.
However, it is very important to remember that when a VUS is discovered in a gene associated with aortic aneurysm conditions, medical genetics providers may recommend that other family members be evaluated with genetic testing and aortic imaging to help determine the importance of the VUS. VUS can be reclassified as pathogenic (harmful or disease-causing) or benign as more information becomes available over time.
HTAD evaluation and genetic testing should be performed by medical specialists familiar with these conditions. This includes medical genetics providers (geneticists, genetic counselors), as well as pediatric and adult cardiologists and cardiothoracic and vascular surgeons.